Die Sichelzellkrankheit (sickle cell disease, SCD) ist eine erblich bedingte schwere Blutkrankheit, bei der die roten Blutkörperchen eine sichelförmige Gestalt annehmen. Sie begleitet Patient*innen ein Leben lang und kann bereits im Kleinkindalter zu zahlreichen ernsten gesundheitlichen Problemen führen.
Im Körper eines Erwachsenen zirkulieren etwa vier bis sechs Liter Blut. Die genaue Menge kann je nach Körpergröße, Geschlecht und Gesundheitszustand variieren. Die rote Farbe des Blutes entsteht durch das eisenhaltige Hämoglobin, welches in den roten Blutkörperchen (Erythrozyten) enthalten ist. Diese Blutzellen haben als Hauptaufgabe, den Körper mit Sauerstoff zu versorgen. Das geschieht, indem Hämoglobin Sauerstoff bindet und die roten Blutkörperchen damit anreichert.1
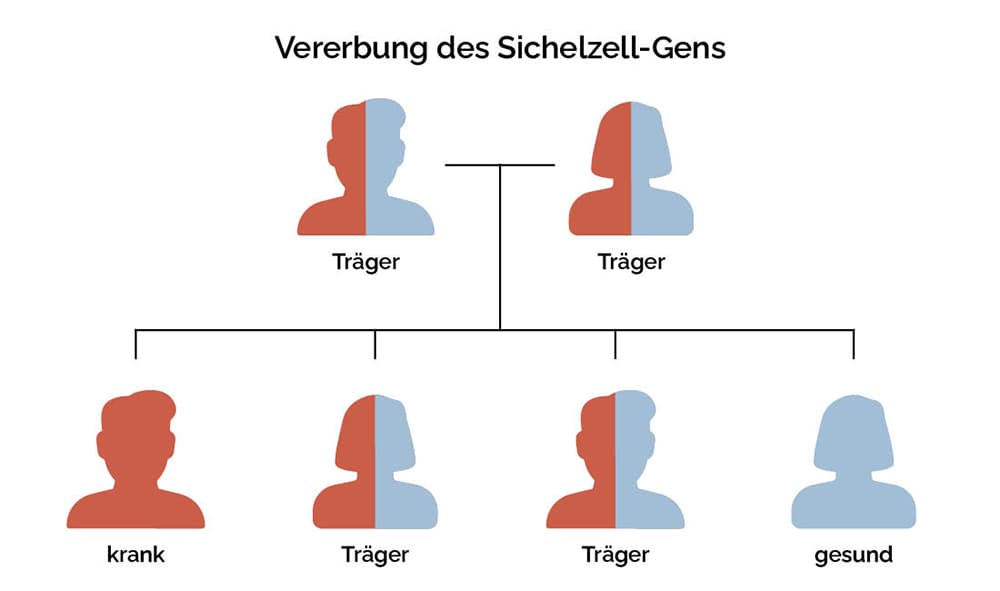
Gesunde rote Blutkörperchen haben die Form einer Scheibe, sind glatt, weich und beweglich. Diese Eigenschaft ermöglicht es ihnen, selbst die kleinsten Blutgefäße zu erreichen. Bei der Sichelzellkrankheit ist das Hämoglobin aufgrund eines Gendefekts krankhaft verändert.2 Die genetische Mutation führt zu einer verminderten Löslichkeit und Fibrillenbildung des Blutfarbstoffs. Dadurch verformen sich die Blutkörperchen und werden sichelförmig, also schmal und spitz.3,4 Sie verlieren ihre Elastizität und leben auch deutlich kürzer (10-20 statt 120 Tage) .5
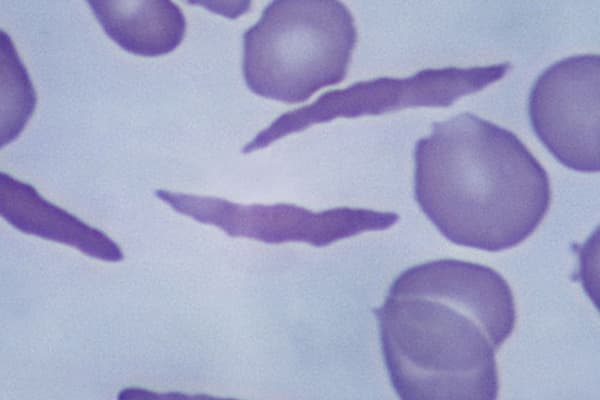
Durch die Verformung und den Elastizitätsverlust passen die veränderten Blutkörperchen nicht mehr so gut durch die kleinen Blutgefäße und können sie verstopfen, wodurch Organe geschädigt werden.
Durch das vorzeitige Absterben der roten Blutkörperchen ist der Körper nicht ausreichend mit Sauerstoff versorgt. Die Folge ist eine chronische Blutarmut, eine so genannte hämolytische Anämie. Die Patient*innen sind blass, sehr müde und erschöpft, antriebslos sowie reizbar. Die Anämie kann auch akut lebensbedrohlich werden, etwa wenn das Blut in der sich plötzlich vergrößernden Milz „versackt“ (akute Milzsequestration). Bei Kindern kann es zu einer Wachstumsverzögerung und verspäteten Pubertät kommen.5,6
Sichelzellen können sich zudem auch auflösen (medizinisch: Hämolyse) und den Inhalt der Blutkörperchen in das umliegende Blutplasma freigeben, was zu Entzündungen, Blutgerinnsel, Blutarmut und weiteren Komplikationen führen kann.
Die Sauerstoffunterversorgung, Gefäßverschlüsse und Entzündungsprozesse sind verantwortlich für viele weitere mitunter schwerwiegende Komplikationen der Sichelzellerkrankung. Diese können unter anderem die Zerstörung von Gewebe durch Mikroinfarkte, schwerste Multiorganerkrankungen, Beingeschwüre, Hauterkrankungen, Schlaganfall und Schmerzkrisen beinhalten.4,7
Erste Symptome von SCD können bereits etwa ab dem 3.-4. Lebensmonat auftreten. Diese beinhalten unter anderem: